Research
Most of the eukaryotic genome is transcribed, yielding a complex repertoire of RNAs that includes tens of thousands of noncoding RNAs with little or no predicted protein-coding capacity. Among these are well-studied small RNAs, such as microRNAs, as well as many other classes of small and long transcripts whose functions and mechanisms of biogenesis are less clear – but likely no less important, especially since some are associated with diseases such as cancer and developmental disorders.
Our goal is to characterize the mechanisms by which these poorly characterized noncoding RNAs are generated, regulated, and function, thereby revealing novel insights into RNA biology and developing new methods to treat diseases.
(1) Circular RNAs: Unexpected outputs of protein-coding genes
Deep sequencing has revealed thousands of eukaryotic protein-coding genes that defy the central dogma, producing circular noncoding RNAs rather than linear messenger RNAs. For some genes, the abundance of the circular RNA exceeds that of the associated linear mRNA by a factor of 10, raising the interesting possibility that the function of some protein-coding genes may actually be to produce circular noncoding RNAs, not proteins. These circular RNAs are generated when the pre-mRNA splicing machinery “backsplices” and joins a splice donor to an upstream splice acceptor (Figure 1). Once produced, it is largely unclear what circular RNAs do, although two are known to efficiently modulate the activity of microRNAs.

Figure 1. Backsplicing produces circular RNAs
From Wilusz and Sharp (2013) Science 340: 440-441.
With the exception of the first and last exons of genes, every other exon in the genome has splicing signals at its 5’ and 3’ ends and theoretically can circularize. However, every exon does not circularize, and, in some cases, multiple exons are present in a circular RNA. We showed that repetitive elements, e.g. SINE elements, in the flanking introns are critical determinants of whether the intervening exon(s) circularize. When repeat sequences from the flanking introns base pair to one another, the splice sites are brought into close proximity and backsplicing occurs. Although mechanistically simple, this step occurs in a highly selective manner, as the sequence of the repeats can drastically alter the efficiency of circular RNA production.
How then is the ratio of linear to circular RNA controlled or modulated? Using RNAi screening in Drosophila cells, we identified many hnRNPs, SR proteins, core spliceosome, and transcription termination factors that control the outputs of reporter and endogenous genes. Surprisingly, when spliceosome components were depleted or inhibited pharmacologically, the steady-state levels of circular RNAs increased while expression of their associated linear mRNAs concomitantly decreased. We propose that this is because cross-exon interactions are not easily replaced with cross-intron interactions, thereby causing spliceosomes to preferentially assemble across an exon and generate a circular RNA. Upon inhibiting RNA polymerase II termination, circular RNA levels were similarly increased due to readthrough transcripts extending into downstream genes and being subjected to backsplicing. In total, these results indicate that inhibition or slowing of canonical pre-mRNA processing events shifts the steady-state output of protein-coding genes towards circular RNAs, which likely helps explain why and how circular RNAs show tissue-specific expression profiles.
Most mature circular RNAs accumulate in the cytoplasm, and we revealed the first insights into how their nuclear export is controlled in a length-dependent manner. We further developed improved methods for circular RNA identification and purification using RNase R by identifying a way to better remove linear RNAs containing G-quadruplexes or structured 3’ ends.
By revealing the fundamental mechanisms by which circular RNAs are generated, we have developed plasmid and viral-based methods for ectopically expressing circular RNAs. Almost any sequence can now be efficiently circularized in eukaryotic cells, which has enabled us (and others) to begin to ask how circular RNAs function. More than 150 labs have requested our circular RNA expression plasmids, highlighting our wide impact on this growing field.
We are continuing to elucidate the mechanism by which circular RNAs are produced. In particular, we are interested in determining how cellular cues can alter the ratio of linear mRNA to circular RNA for a given gene. We are also focused on identifying biological functions for circular RNAs, thereby revealing novel insights into how circular RNAs fit into the regulatory landscape of the cell.
KEY PUBLICATIONS
Dodbele, S., Mutlu, N., and Wilusz, J.E. (2021) Best practices to ensure robust investigation of circular RNAs: pitfalls and tips. EMBO Rep 22: e52072.
Xiao, M.S. and Wilusz, J.E. (2019) An improved method for circular RNA purification using RNase R that efficiently removes linear RNAs containing G-quadruplexes or structured 3’ ends. Nucleic Acids Res 47: 8755-8769.
Huang, C., Liang, D., Tatomer, D.C., and Wilusz, J.E. (2018) A length-dependent evolutionarily conserved pathway controls nuclear export of circular RNAs. Genes Dev 32: 639-644.
Liang, D., Tatomer, D.C., Luo, Z., Wu, H., Yang, L., Chen, L.L., Cherry, S., and Wilusz, J.E. (2017) The output of protein-coding genes shifts to circular RNAs when the pre-mRNA processing machinery is limiting. Mol Cell 68: 940-954.
Tatomer, D.C., Liang, D., and Wilusz, J.E. (2017) Inducible expression of eukaryotic circular RNAs from plasmids. Methods Mol Biol 1648: 143-154.
Chen, Y.G., Kim, M.V., Chen, X., Batista, P.J., Aoyama, S., Wilusz, J.E., Iwasaki, A., and Chang, H.Y. (2017) Sensing self and foreign circular RNAs by intron identity. Mol Cell 67: 228-238.
Kramer, M.C., Liang, D., Tatomer, D.C., Gold, B., March, Z.M., Cherry, S., and Wilusz, J.E. (2015) Combinatorial control of Drosophila circular RNA expression by intronic repeats, hnRNPs, and SR proteins. Genes Dev 29: 2168-2182.
Liang, D. and Wilusz, J.E. (2014) Short intronic repeat sequences facilitate circular RNA production. Genes Dev 28: 2233-2247.
Wilusz, J.E. and Sharp, P.A. (2013) A circuitous route to noncoding RNA. Science 340: 440-441.
(2) The Integrator complex cleaves nascent mRNAs to attenuate transcription
Cellular homeostasis requires transcriptional outputs to be coordinated, and many events post transcription initiation can dictate the levels and functions of mature transcripts. To systematically identify regulators of inducible gene expression, we performed high-throughput RNAi screening of the Drosophila Metallothionein A (MtnA) promoter. This surprisingly revealed that the Integrator complex, which has a well-established role in 3' end processing of small nuclear RNAs (snRNAs), attenuates MtnA transcription during copper stress. Integrator complex subunit 11 (IntS11) endonucleolytically cleaves MtnA transcripts, resulting in premature transcription termination and degradation of the nascent RNAs by the RNA exosome, a complex also identified in the screen (Figure 2). Using RNA-seq, we then identified >400 additional Drosophila protein-coding genes whose expression increases upon Integrator depletion. We focused on a subset of these genes and confirmed that Integrator is bound to their 5' ends and negatively regulates their transcription via IntS11 endonuclease activity. In fact, Integrator catalyzed premature transcription termination events can repress the expression of some full-length mRNAs by more than 100-fold.
Many non-catalytic Integrator subunits, which are largely dispensable for snRNA processing, also have regulatory roles at these protein-coding genes, possibly by controlling Integrator recruitment or RNA polymerase II dynamics. Altogether, our results suggest that attenuation via Integrator cleavage limits production of many full-length mRNAs, allowing precise control of transcription outputs. Going forward, it will be highly interesting to understand how the Integrator complex is recruited and regulated as well as why mutations in Integrator subunits are associated with human diseases.
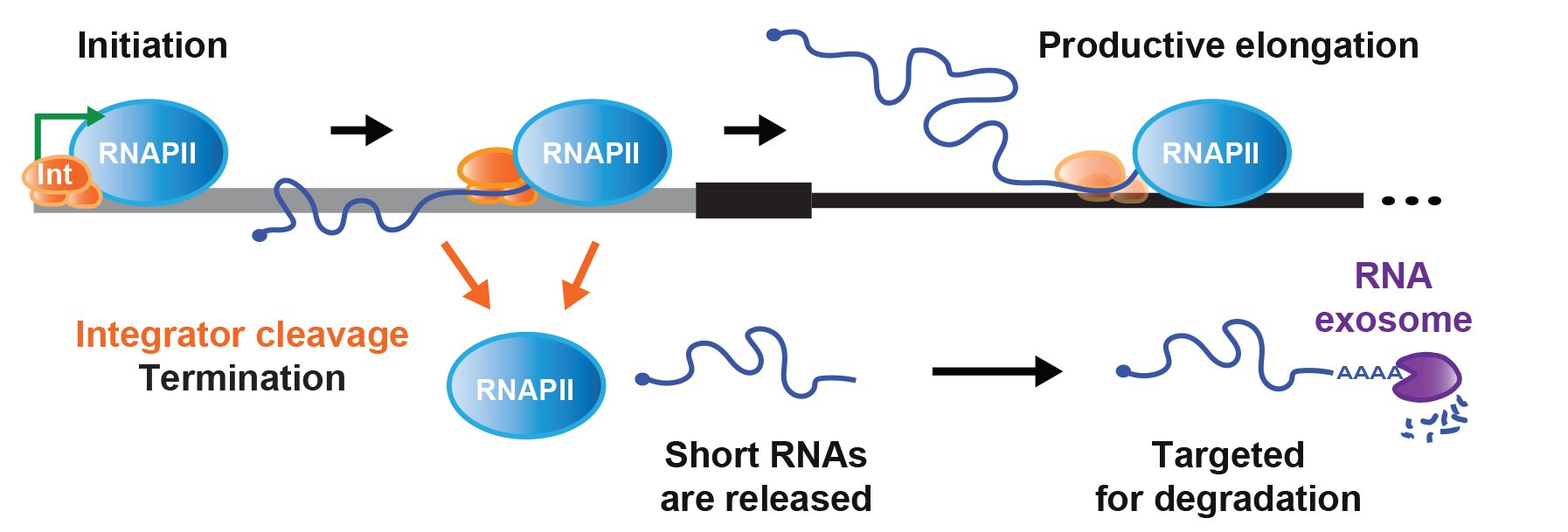
Figure 2. Integrator cleavage attenuates protein-coding gene expression
From Tatomer et al. (2019) Genes Dev 33: 1525-1538.
KEY PUBLICATIONS
Fujiwara, R., Zhai, S.-N., Liang, D., Shah, A.P., Tracey, M., Ma, X.-K., Fields, C.J., Mendoza-Figueroa, M.S., Meline, M.C., Tatomer, D.C., Yang, L., and Wilusz, J.E. (2023) IntS6 and the Integrator phosphatase module tune the efficiency of select premature transcription termination events. Mol Cell 83: 4445-4460..
Mendoza-Figueroa, M.S., Tatomer, D.C., and Wilusz, J.E. (2020) The Integrator complex in transcription and development. Trends Biochem Sci 45: 923-934.
Tatomer, D.C. and Wilusz, J.E. (2020) Attenuation of eukaryotic protein-coding gene expression via premature transcription termination. Cold Spring Harb Symp Quant Biol 84: 83-93.
Elrod, N.D., Henriques, T., Huang, K.L., Tatomer, D.C., Wilusz, J.E., Wagner, E.J., and Adelman, K. (2019) The Integrator complex attenuates promoter-proximal transcription at protein-coding genes. Mol Cell 76:738-752.
Tatomer, D.C., Elrod, N.D., Liang, D., Xiao, M.S., Jiang, J.Z., Jonathan, M., Huang, K.L., Wagner, E.J., Cherry, S., and Wilusz, J.E. (2019) The Integrator complex cleaves nascent mRNAs to attenuate transcription. Genes Dev 33: 1525-1538.
(3) Regulation of non-AUG translation by ribosome queuing
Aberrant translation initiation at non-AUG start codons is associated with cancer and neurodegenerative diseases. Therefore, identifying how non-AUG translation is regulated differently from canonical translation may reveal new therapeutic opportunities. We showed that non-AUG translation is strikingly resistant to multiple protein synthesis inhibitors, including cycloheximide. We find that these inhibitors slow elongating ribosomes, thereby inducing a queue of pre-initiation complexes that can become positioned over and initiate from an otherwise poorly recognized start codon (Figure 3). This work has important implications for how translation of medically important genes occurs (e.g. RAN translation in neurodegenerative diseases) and how to pharmacologically modulate these events. Going forward, it will be highly interesting to develop strategies that block ribosome queues, in part by revealing the proteins that control their formation and dynamics.

Figure 3. Ribosome queuing enables increased non-AUG translation
From Kearse et al. (2019) Genes Dev 33: 871-885.
KEY PUBLICATIONS
Kearse, M.G., Goldman, D.H., Choi, J., Nwaezeapu, C., Liang, D., Green, K.M., Goldstrohm, A.C., Todd, P.K., Green, R., and Wilusz, J.E. (2019) Ribosome queuing enables non-AUG translation to be resistant to multiple protein synthesis inhibitors. Genes Dev. 33: 871-885.
Kearse, M.G. and Wilusz, J.E. (2017) Non-AUG translation: a new start for protein synthesis in eukaryotes. Genes Dev. 31: 1717-1731.
(4) Regulation of MALAT1, MEN β, and other non-polyadenylated RNAs
Much of our early work focused on the MALAT1 locus, which is over-expressed in many human cancers and produces a long nuclear-retained noncoding RNA as well as a tRNA-like small RNA known as mascRNA, MALAT1-associated small cytoplasmic RNA. Despite being an RNA polymerase II transcript, the 3’ end of MALAT1 is produced not by canonical cleavage/polyadenylation but instead by recognition and cleavage of the tRNA-like structure by RNase P (Figure 4).

Figure 4. Non-canonical 3’ end processing of MALAT1
The MEN β long nuclear-retained noncoding RNA, also known as NEAT1_2, is similarly processed at its 3’ end by RNase P. Surprisingly, although processing of MEN β produces a tRNA-like small RNA, it is generally rapidly degraded in vivo. Pursuing this observation, we revealed a universally conserved quality control mechanism mediated by the CCA-adding enzyme. Normally, the CCA-adding enzyme adds CCA to the 3’ ends of tRNAs, a critical step in tRNA biogenesis that generates the amino acid attachment site. However, the enzyme adds CCACCA to structurally unstable tRNAs and tRNA-like small RNAs (including the MEN β tRNA-like transcript and certain hypomodified tRNAs) marking them for degradation. We conjecture that CCACCA addition prevents errors in translation and controls tRNA levels, an especially critical function considering that even slight changes in tRNA levels can drive cell proliferation and oncogenic transformation.
As the mature 3’ ends of MALAT1 and MEN β are generated by RNase P and not by the cleavage and polyadenylation machinery, these long noncoding RNAs lack poly(A) tails. Nevertheless, these RNAs are expressed at levels higher than many protein-coding genes. We showed that the 3’ ends of these noncoding RNAs are protected from 3’-5’ exonucleases by highly conserved triple helical structures. Surprisingly, when these triple helical structures are placed downstream from an open reading frame, the transcript is efficiently translated in vivo despite the lack of a poly(A) tail. This result challenges the common paradigm that long poly(A) tails are required for RNA stability and efficient protein synthesis and suggests that non-polyadenylated RNAs may produce functional peptides in vivo via mechanisms that are likely independent of poly(A) binding protein.
KEY PUBLICATIONS
Doucet, A.J., Wilusz, J.E., Miyoshi, T., Liu, Y., and Moran, J.V. (2015) A 3’ poly(A) tract is required for LINE-1 retrotransposition. Mol Cell 60: 728-741
Kuhn, C.-D., Wilusz, J.E., Zheng, Y., Beal, P.A., and Joshua-Tor, L. (2015) On-enzyme refolding permits small RNA and tRNA surveillance by the CCA-adding enzyme. Cell 160: 644-658.
Wilusz, J.E., JnBaptiste, C.K., Lu, L.Y., Kuhn, C.-D., Joshua-Tor, L., and Sharp, P.A. (2012) A triple helix stabilizes the 3’ ends of long noncoding RNAs that lack poly(A) tails. Genes Dev 26: 2392-2407.
Wilusz, J.E., Whipple, J.M., Phizicky, E.M., and Sharp, P.A. (2011) tRNAs marked with CCACCA are targeted for degradation. Science 334: 817-821.
Wilusz, J.E. and Spector, D.L. (2010) An unexpected ending: non-canonical 3’ end processing mechanisms. RNA 16: 259-266.
Sunwoo, H., Dinger, M.E., Wilusz, J.E., Amaral, P.P., Mattick, J.S., and Spector, D.L. (2009) MEN ε/β nuclear-retained non-coding RNAs are up-regulated upon muscle differentiation and are essential components of paraspeckles. Genome Res 19: 347-359.
Wilusz, J.E., Freier, S.M., and Spector, D.L. (2008) 3’ end processing of a long nuclear-retained noncoding RNA yields a tRNA-like cytoplasmic RNA. Cell 135: 919-932.